Fibrosi cistica
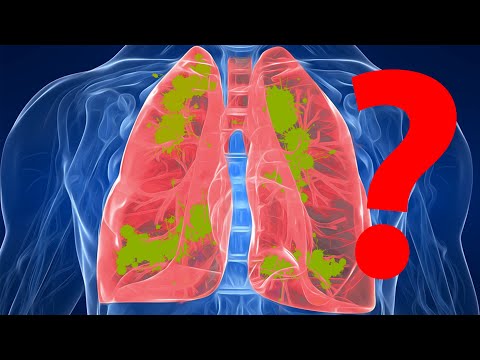
La fibrosi cistica è una malattia che provoca l'accumulo di muco denso e appiccicoso nei polmoni, nel tratto digestivo e in altre aree del corpo. È una delle malattie polmonari croniche più comuni nei bambini e nei giovani adulti. È una malattia pericolosa per la vita.
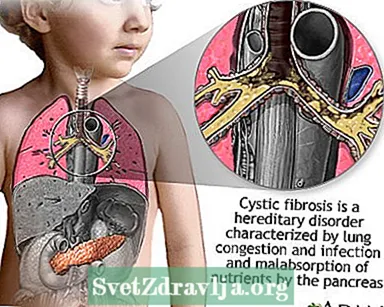
La fibrosi cistica (FC) è una malattia che si trasmette attraverso le famiglie. È causato da un gene difettoso che fa produrre al corpo un fluido anormalmente denso e appiccicoso, chiamato muco. Questo muco si accumula nelle vie respiratorie dei polmoni e nel pancreas.
L'accumulo di muco provoca infezioni polmonari potenzialmente letali e gravi problemi di digestione. La malattia può anche colpire le ghiandole sudoripare e il sistema riproduttivo di un uomo.
Molte persone portano un gene CF, ma non hanno sintomi. Questo perché una persona con FC deve ereditare 2 geni difettosi, 1 da ciascun genitore. Alcuni americani hanno il gene CF. È più comune tra quelli di discendenza dell'Europa settentrionale o centrale.
La maggior parte dei bambini con FC viene diagnosticata all'età di 2 anni, soprattutto perché lo screening neonatale viene eseguito negli Stati Uniti. Per un piccolo numero, la malattia non viene rilevata fino all'età di 18 anni o più. Questi bambini hanno spesso una forma più lieve della malattia.
I sintomi nei neonati possono includere:
- Crescita ritardata
- Mancato aumento di peso normalmente durante l'infanzia
- Nessun movimento intestinale nelle prime 24-48 ore di vita
- Pelle dal sapore salato
I sintomi legati alla funzione intestinale possono includere:
- Dolore alla pancia da stitichezza grave
- Aumento di gas, gonfiore o pancia che appare gonfia (dilatata)
- Nausea e perdita di appetito
- Feci pallide o color argilla, maleodoranti, con muco o che galleggiano
- Perdita di peso
I sintomi relativi ai polmoni e ai seni paranasali possono includere:
- Tosse o aumento del muco nei seni o nei polmoni
- Fatica
- Congestione nasale causata da polipi nasali
- Episodi ripetuti di polmonite (i sintomi di polmonite in qualcuno con fibrosi cistica includono febbre, aumento della tosse e mancanza di respiro, aumento del muco e perdita di appetito)
- Dolore al seno o pressione causata da infezione o polipi
Sintomi che possono essere notati più tardi nella vita:
- Infertilità (negli uomini)
- Infiammazione ripetuta del pancreas (pancreatite)
- Sintomi respiratori
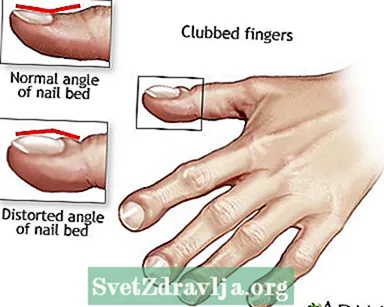
- dita bastonate
Viene eseguito un esame del sangue per aiutare a rilevare la fibrosi cistica. Il test cerca cambiamenti nel gene CF. Altri test utilizzati per diagnosticare la FC includono:
- Il test immunoreattivo del tripsinogeno (IRT) è un test standard di screening neonatale per la fibrosi cistica. Un alto livello di IRT suggerisce una possibile FC e richiede ulteriori test.
- Il test del cloruro del sudore è il test diagnostico standard per la fibrosi cistica. Un alto livello di sale nel sudore della persona è un segno della malattia.
Altri test che identificano problemi che possono essere correlati alla FC includono:
- Radiografia del torace o TAC
- Test del grasso fecale
- Test di funzionalità polmonare
- Misurazione della funzione pancreatica (elastasi pancreatica fecale)
- Test di stimolazione della secretina
- Tripsina e chimotripsina nelle feci
- Serie GI superiore e intestino tenue
- Colture polmonari (ottenute mediante espettorato, broncoscopia o tampone faringeo)
Una diagnosi precoce della FC e un piano di trattamento possono migliorare sia la sopravvivenza che la qualità della vita. Il follow-up e il monitoraggio sono molto importanti. Quando possibile, le cure dovrebbero essere ricevute presso una clinica specializzata in fibrosi cistica. Quando i bambini raggiungono l'età adulta, dovrebbero trasferirsi in un centro specializzato in fibrosi cistica per adulti.
Il trattamento per i problemi polmonari include:
- Antibiotici per prevenire e curare le infezioni polmonari e dei seni paranasali. Possono essere assunti per via orale o somministrati nelle vene o mediante trattamenti respiratori. Le persone con FC possono assumere antibiotici solo quando necessario o sempre. Le dosi sono spesso più alte del normale.
- Medicinali per inalazione per aiutare ad aprire le vie aeree.
- Altri medicinali che vengono somministrati da un trattamento respiratorio per fluidificare il muco e facilitare la tosse sono la terapia enzimatica della DNAsi e soluzioni saline altamente concentrate (soluzione salina ipertonica).
- Vaccino antinfluenzale e vaccino pneumococcico polisaccaridico (PPV) ogni anno (chiedi al tuo medico).
- Il trapianto di polmone è un'opzione in alcuni casi.
- L'ossigenoterapia può essere necessaria quando la malattia polmonare peggiora.
I problemi polmonari vengono trattati anche con terapie per fluidificare il muco. Questo rende più facile tossire il muco dai polmoni.
Questi metodi includono:
- Attività o esercizio che ti fa respirare profondamente
- Dispositivi che vengono utilizzati durante il giorno per aiutare a liberare le vie aeree da troppo muco
- Percussione toracica manuale (o fisioterapia toracica), in cui un membro della famiglia o un terapista batte leggermente il petto, la schiena e l'area sotto le braccia della persona
Il trattamento per problemi intestinali e nutrizionali può includere:
- Una dieta speciale ricca di proteine e calorie per bambini più grandi e adulti
- Enzimi pancreatici per aiutare ad assorbire grassi e proteine, che vengono assunti ad ogni pasto
- Integratori vitaminici, in particolare vitamine A, D, E e K
- Il tuo medico può consigliare altri trattamenti se hai feci molto dure
Ivacaftor, lumacaftor, tezacaftor ed elexacaftor sono medicinali che trattano alcuni tipi di FC.
- Migliorano la funzione di uno dei geni difettosi che causa la fibrosi cistica.
- Fino al 90% dei pazienti con FC e idonei per uno o più di questi medicinali da soli o in combinazione.
- Di conseguenza, c'è meno accumulo di muco denso nei polmoni. Anche altri sintomi della FC sono migliorati.
L'assistenza e il monitoraggio domiciliare dovrebbero includere:
- Evitare fumo, polvere, sporco, fumi, prodotti chimici domestici, fumo di camino e muffe.
- Somministrare molti liquidi, specialmente a neonati e bambini quando fa caldo, quando c'è diarrea o feci molli, o durante un'attività fisica extra.
- Esercizio 2 o 3 volte a settimana. Nuoto, jogging e ciclismo sono buone opzioni.
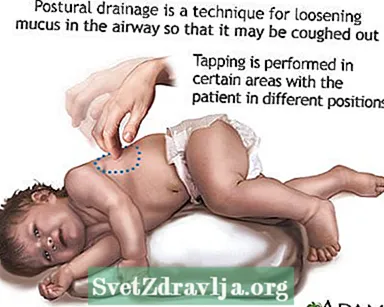
- Eliminare o sollevare muco o secrezioni dalle vie aeree. Questo deve essere fatto da 1 a 4 volte al giorno. I pazienti, le famiglie e gli operatori sanitari devono imparare a eseguire le percussioni toraciche e il drenaggio posturale per aiutare a mantenere libere le vie aeree.
- Non è raccomandato alcun contatto con altre persone con FC in quanto possono scambiare infezioni (non si applica ai membri della famiglia).
Puoi alleviare lo stress della malattia unendoti a un gruppo di supporto per la fibrosi cistica. Condividere con altri che hanno esperienze e problemi comuni può aiutare la tua famiglia a non sentirsi sola.
La maggior parte dei bambini con FC rimane in buona salute fino al raggiungimento dell'età adulta. Possono prendere parte alla maggior parte delle attività e frequentare la scuola. Molti giovani adulti con FC finiscono l'università o trovano lavoro.
La malattia polmonare alla fine peggiora fino al punto in cui la persona è disabile. Oggi, la durata media della vita delle persone con FC che vivono fino all'età adulta è di circa 44 anni.
La morte è più spesso causata da complicazioni polmonari.
La complicanza più comune è l'infezione respiratoria cronica.
Altre complicazioni includono:
- Problemi intestinali, come calcoli biliari, blocco intestinale e prolasso rettale
- Tossendo sangue
- Insufficienza respiratoria cronica
- Diabete
- infertilità
- Malattia epatica o insufficienza epatica, pancreatite, cirrosi biliare
- Malnutrizione
- Polipi nasali e sinusite
- Osteoporosi e artrite
- Polmonite che continua a tornare
- Pneumotorace
- Insufficienza cardiaca destra (cuore polmonare)
- Cancro colorettale
Chiama il tuo fornitore se un neonato o un bambino ha sintomi di FC e sperimenta:
- Febbre, aumento della tosse, alterazioni dell'espettorato o del sangue nell'espettorato, perdita di appetito o altri segni di polmonite
- Aumento della perdita di peso
- Movimenti intestinali più frequenti o feci maleodoranti o con più muco
- Pancia gonfia o gonfiore aumentato
Chiama il tuo medico se una persona con FC sviluppa nuovi sintomi o se i sintomi peggiorano, difficoltà respiratorie particolarmente gravi o tosse con sangue.
La CF non può essere prevenuta. Lo screening di quelli con una storia familiare della malattia può rilevare il gene CF in molti portatori.
CF
- Nutrizione enterale - bambino - gestione dei problemi
- Tubo di alimentazione gastrostomico - bolo
- Come respirare quando si è a corto di fiato
- Sonda per digiunostomia
- Drenaggio posturale
 discoteca
discoteca Drenaggio posturale
Drenaggio posturale dita bastonate
dita bastonate Fibrosi cistica
Fibrosi cistica
Donaldson SH, Pilewski JM, Griese M, et al. Tezacaftor/ivacaftor in soggetti con fibrosi cistica e F508del/F508del-CFTR o F508del/G551D-CFTR. Am J Respir Crit Care Med. 2018;197(2):214-224. PMID: 28930490 pubmed.ncbi.nlm.nih.gov/28930490/.
Eagan ME, Schechter MS, Voynow JA. Fibrosi cistica. In: Kliegman RM, St. Geme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM, eds. Nelson Textbook of Pediatrics. 21a ed. Filadelfia, PA: Elsevier; 2020: cap 432.
Farrell PM, White TB, Ren CL, et al. Diagnosi della fibrosi cistica: linee guida di consenso della Cystic Fibrosis Foundation. J Pediatra. 2017;181S:S4-S15.e1. PMID: 28129811 pubmed.ncbi.nlm.nih.gov/28129811/.
Graeber SY, Dopfer C, Naehrlich L, et al. Effetti della terapia con lumacaftor/ivacaftor sulla funzione CFTR in pazienti omozigoti Phe508del con fibrosi cistica. Am J Respir Crit Care Med. 2018;197(11):1433-1442. PMID: 29327948 pubmed.ncbi.nlm.nih.gov/29327948/.
Grasemann H. Fibrosi cistica. In: Goldman L, Schafer AI, eds. Medicina Goldman-Cecil. 26a ed. Filadelfia, PA: Elsevier; 2020: cap 83.
Rowe SM, Hoover W, Solomon GM, Sorscher EJ. Fibrosi cistica. In: Broaddus VC, Mason RJ, Ernst JD, et al, eds. Il libro di testo di medicina respiratoria di Murray e Nadel. 6a ed. Filadelfia, PA: Elsevier Saunders; 2016: cap 47.
Taylor-Cousar JL, Munck A, McKone EF, et al. Tezacaftor-ivacaftor in pazienti con fibrosi cistica omozigoti per phe508del. N Inglese J Med. 2017;377(21):2013-2023. PMID: 29099344 pubmed.ncbi.nlm.nih.gov/29099344/.
Articoli Per Te

Cos'è la terapia Bowen?
